

I thought I’d take a moment to discuss how exactly biologists create phylogenetic trees. Our discussion will not include factors such as morphological comparisons etc.- which was more widely used before genetics came along- but rather the molecular methods used today. I thought this would be better served as a blog post rather than a podcast episode, as in blog format I can use images to illustrate what I’m saying (which is predictably rather difficult in audio format!).
So, let’s say that you’re an ecologist who’s just discovered an uninhabited island far out to sea. There’s three species of snake on this island (hence the picture at the top) and you want to know how long ago these species diverged from their common ancestor. You find a fossil which tells you that species A and species C must have diverged 15 million years ago, but you can’t find anything for the other species. So, instead, you sequence a protein common to all species (let’s say haemoglobin for the sake of argument) which gives you the results in Table 1. How does this help you?
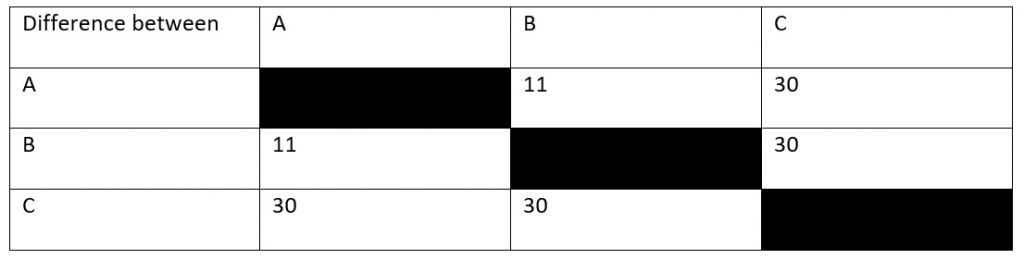
Looking at our table, you can see that, in 15 million years, there have been thirty amino acid sequence changes between species A and species C. However, this doesn’t quite give us our rate of mutation yet. To know why this is the case, you’ve got to consider that both species have been evolving away from their common origin. This means that we’ve effectively got to double the time between them to 30 million years. After all, species A has been evolving for 15 million years and species C has been.
This means that, if we assume a constant mutation rate, we end up with a rate of 1 mutation per million years. What we’ve done is calibrate our molecular clock- we now know the rate of mutation for the protein we’re using to build our tree.
So, let’s now look at the difference between species B and species C, which is also 30 mutations. This means that these two species also had a total evolution time of thirty million years. Dividing by two, we get a divergence time of 15 million years, as with species A. By contrast, species A and species B have eleven amino acid differences, meaning that they must have diverged 5.5 million years ago.
How does all of this fit together? Well, A and B must have diverged after both the A-C divergence or the B-C divergence. This means that C must have split off from the common ancestor first, and then A and B diverged, as shown below:
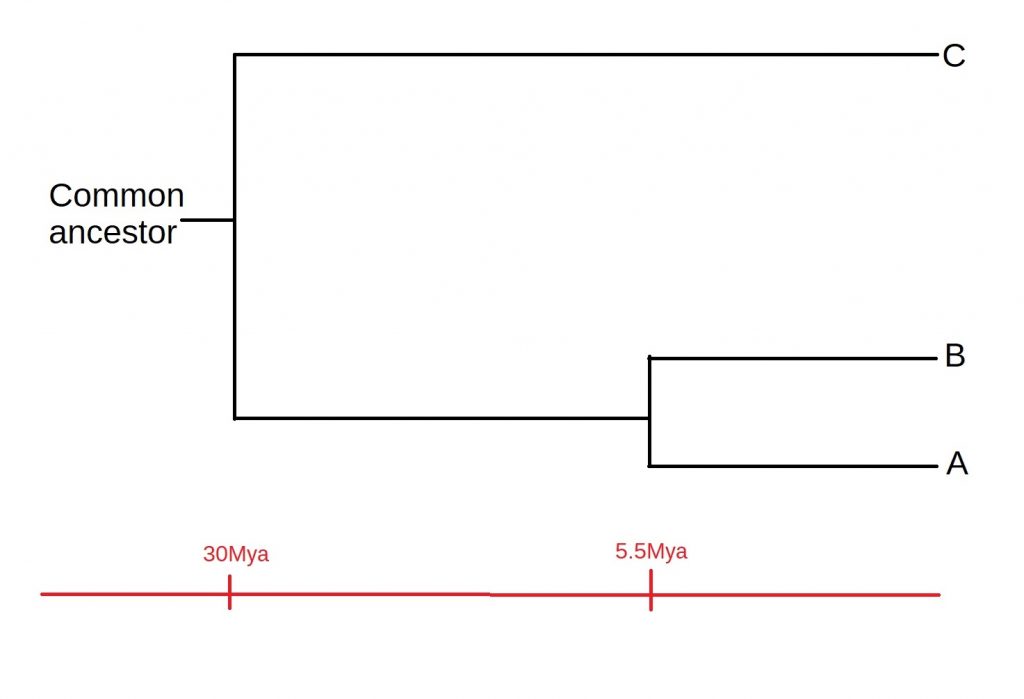
Of course, there are some assumptions we’ve made while constructing our tree, such as a constant rate of mutation. Added to this, most phylogenies will be a lot more complicated than just three species. However, it serves well as an example of the process by which such tree are constructed. A more in-depth discussion of these assumptions, as well as the process of speciation itself, is a story for another time.
- Image Credit:
- Image: Pixabay
- User: Syaibatulhamdi
- Featured image: Mountains Photographer Man – Free image on Pixabay
- Image labelled for reuse
- No changes have been made